

I virus, sono dei microrganismi con una capacità replicativa “esplosiva”, in quanto in ogni ciclo di infezione all’interno della cellula ospite da un unico virus si generano non due, ma milioni di esemplari di progenie. Questa enorme capacità replicativa li rende molto plastici nei confronti dell’ambiente in cui si riproducono, nel senso che la velocità di evoluzione è molto accelerata e ne determina il rapido adattamento a nicchie ecologiche nuove o mutevoli. Questa caratteristica è legata al fatto che in ogni ciclo replicativo l’enzima che genera le copie seriali del materiale genetico del virus commette occasionalmente degli errori di copiatura. Questi errori di copiatura nel genoma virale costituiscono le mutazioni, che a loro volta determinano il cambiamento della sequenza aminoacidica delle proteine virali codificate dalli geni mutati.
Le mutazioni in genere alterano l’equilibrio del virus nella sua nicchia naturale, e quindi il più delle volte hanno un effetto negativo sulla sua riproduzione, per cui le particelle virali mutate spariscono dalla circolazione. Raramente qualche mutazione conferisce al virus una migliore capacità di replicarsi in determinate condizioni, e quindi si fissano nella popolazione virale, che, accumulando mutazioni generazione dopo generazione, evolve adattandosi a nuovi ospiti e a nuovi habitat.
E’ questa capacità all’origine del salto di specie dei virus, anche detto “spill over”, che si verifica ogni volta che un virus passa accidentalmente dalla specie animale che rappresenta il suo ospite naturale all’uomo e, accumulando mutazioni su mutazioni, acquisisce la capacità di trasmettersi con efficienza fra gli esseri umani. All’interno della specie umana questo processo di adattamento continua, e può essere tracciato mediante il sequenziamento sistematico dei genomi.
I virus a DNA sono meno proni ad accumulare mutazioni, perché l’enzima DNA polimerasi DNA-dipendente, che replica il loro genoma è un enzima replicativo relativamente fedele, che commette pochi errori di copiatura, mentre i virus a RNA presentano una variabilità genomica molto più accentuata, perché la vasta gamma di enzimi che copiano il genoma a RNA (la famiglia delle RNA polimerasi RNA-dipendenti) è caratterizzata da scarsa fedeltà.
Il SARS-CoV-2, che è l’agente virale responsabile della sindrome denominata COVID-19, appartiene alla famiglia dei Coronaviridae, caratterizzata da un genoma a RNA a singola elica, polarità positiva, che codifica per più di 20 proteine necessarie alla replicazione (proteine non strutturali e accessorie) e per 4 proteine strutturali, che compongono la particella virale. Tra le proteine che compongono la particella virale quello situata sull’involucro esterno, denominata glicoproteina di superficie, o S (Spike), è responsabile, attraverso una regione denominata RBD (Receptor Binding Domain), dell’aggancio del virus al recettore situato sulla superficie della cellula bersaglio, ACE2, e quindi del suo ingresso nella cellula che ospiterà la replicazione virale. Dopo l’aggancio del recettore, la glicoproteina subisce un riarrangiamento che provoca la fusione della membrana del virus con la membrana della cellula ospite. La glicoproteina Spike è quindi responsabile delle prime fasi della infezione, ed è proprio contro di essa che sono rivolti gli anticorpi che bloccano l’infezione nelle fasi più precoci. È per questo motivo che i vaccini contro il SARS-CoV-2 sono basati sulla glicoproteina S.
Fra i virus a RNA, la variabilità del SARS-CoV-2 è decisamente inferiore a quella di altri virus, quali il virus dell’epatite C, il virus dell’immunodeficienza umana acquisita (HIV) e il virus influenzale, ma comunque è notevole, determinando un accumulo di mutazioni che si fissano al ritmo di un paio al mese. Mentre la maggior parte delle mutazioni non ha un impatto significativo, qualcuna può conferire al virus caratteristiche favorevoli alla sua diffusione, come una maggiore trasmissibilità o la capacità di sfuggire alle difese immunitarie acquisite per infezione naturale o per vaccinazione (escape immunologico). Le mutazioni di per sé potrebbero anche determinare un aumento di virulenza, con forme cliniche più severe, ma va precisato che maggiore trasmissibilità non è sinonimo di maggiore virulenza o patogenicità. In ogni caso, la maggiore trasmissibilità si traduce comunque in un maggior numero assoluto di infezioni, determinando, così, anche un aumento del numero di casi gravi. Per quanto detto, la variabilità genetica del SARS-CoV-2 è oggetto di intenso monitoraggio, per identificare le varianti che emergono e valutare se vi sono rischi aumentati di trasmissibilità, escape immunologico e patogenicità.
Il database internazionale GISAID (acronimo di Global Initiative on Sharing Avian Influenza Data, https://www.gisaid.org/) inizialmente dedicato all’influenza, e successivamente esteso ad altri virus con potenziale epidemico, quali Ebola, Zika e, in ultimo, SARS-CoV-2, è nato nel 2008 con l’intento di fornire una piattaforma per condividere i dati di sequenze virali. Ad oggi sono depositate in GISAID e disponibili per la comunità scientifica internazionale più di 5 milioni di sequenze del SARS-CoV-2, che hanno permesso di tracciare, fin dagli esordi della pandemia, il percorso evolutivo del virus nel mondo.
Fin dalle prime fasi della pandemia sono comparsi ceppi varianti del SARS-CoV-2 rispetto al ceppo originale emerso a Wuhan in Cina: finora ne sono stati identificati in tutto il mondo centinaia. Già nelle prime fasi della pandemia una mutazione nella posizione 614 della glicoproteina S (G614S) ha determinato un repentino aumento della trasmissibilità del virus, e si è diffusa in maniera dilagante, tanto che oggi quasi tutti i ceppi circolanti contengono questa mutazione. Su questo background si sono aggiunte ulteriori mutazioni, alcune delle quali interessano proprio la regione che lega il recettore (RBD), altre interessano porzioni più esterne di S, coinvolte nel riarrangiamento della glicoproteina che precede la fusione con la membrana cellulare.
L’Organizzazione Mondiale della Sanità (OMS) e la sua rete internazionale di esperti monitorano costantemente le modifiche del virus, in modo che, se vengono identificate mutazioni significative, è possibile allertare i Paesi affinché adottino tempestivamente interventi per prevenire la loro diffusione.
L’OMS e le altre organizzazioni internazionali hanno adottato un sistema per designare le varianti in base al rischio associato con la loro diffusione: Variant of Concern (VOC), Variant of Interest (VOI), o Variant Under Monitoring (VUM). La tipologia di varianti che merita maggiore attenzione è la VOC, che indica varianti le cui mutazioni possono determinare un concreto rischio di aumentata diffusione e possibile evasione dall’immunità. Le VOC sono designate con lettere dell’alfabeto greco.
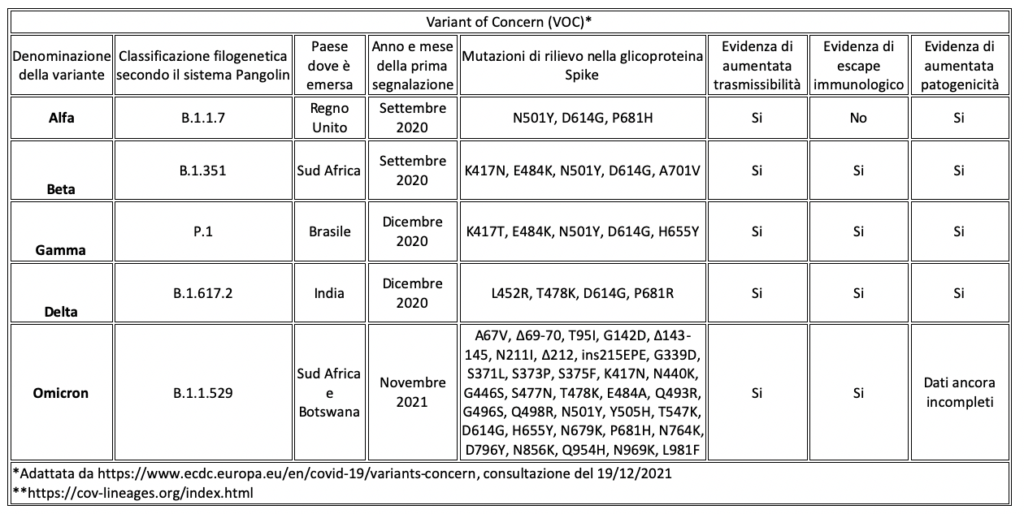
Le principali VOC che si sono succedute negli ultimi mesi, secondo il centro europeo di controllo delle malattie ECDC (https://www.ecdc.europa.eu/en/covid-19/variants-concern) sono riportate nella Tabella, e brevemente descritte di seguito.
- Variante Alfa, identificata per la prima volta nel Regno Unito nel settembre 2020, e diffusasi in maniera esplosiva in tutto il mondo fino a rappresentare la quasi totalità delle infezioni nei primi mesi del 2021. Al momento non è più considerata VOC, in quanto non ha dimostrato rilevanti caratteristiche di virulenza e di evasione immunitaria, ed è stata quasi totalmente soppiantata nella diffusione a livello mondiale dalla variante delta.
- Variante Beta, identificata per la prima volta in Sud Africa a settembre 2020. Non si è diffusa in maniera esplosiva, ma i dati disponibili indicano che, nonostante non sembri caratterizzata da una maggiore trasmissibilità, questa variante potrebbe possedere caratteristiche di parziale escape immunologico, per cui viene monitorata con attenzione.
- Variante Gamma, identificata per la prima volta in Brasile nel dicembre 2020.
Non ha avuto una diffusione esplosiva; gli studi hanno dimostrato una potenziale maggiore trasmissibilità e un possibile rischio di reinfezione. Non sono disponibili evidenze sulla maggiore gravità della malattia. - Variante Delta, rilevata per la prima volta in India nel dicembre 2020.
Questa variante è caratterizzata da una trasmissibilità decisamente più elevata rispetto alla variante Alfa, mostra un periodo di incubazione più breve, ed è associata ad un rischio relativamente più elevato di infezione in soggetti parzialmente vaccinati. - Variante Omicron (rilevata per la prima volta in Sud Africa e Botswana nel novembre 2021. Questa variante presenta un numero elevato di mutazioni del gene S, incluse quasi tutte quelle riscontrate nelle varianti precedenti, e in più ne contiene di nuove, per cui si teme che possa presentare un significativo grado di escape immunologico. Infatti si stanno accumulando dati che indicano che molte infezioni con la variane omicron si verificano anche in soggetti vaccinati o guariti. La rapida diffusione sia in Sud Africa dove è emersa, sia in tutti i paesi nei quali finora è stata rilevata, fa supporre che questa variante abbia un significativo aumento di trasmissibilità rispetto alla delta, che sta rapidamente indietreggiando rispetto a questa nuova variante. Non vi sono ancora dati definitivi sulla virulenza, ma, anche se la patogenicità intrinseca del ceppo non sembra aumentata, la sua rapidità di diffusione fa temere una esplosione di infezioni accompagnata da un rilevante numero assoluto di casi gravi.
Da questo excursus è evidente che l’emergenza dei cambiamenti genomici che determinano la comparsa e la affermazione delle varianti di SARS-CoV-2 rappresentano un rischio per la salute globale e meritano la massima attenzione. Il fenomeno è intrinsecamente connesso con la continua replicazione del virus, e solo arrestandola in maniera drastica si potrà porre un freno all’accumulo di mutazioni. Finché la diffusione del virus non sarà arginata da un’adeguata copertura immunitaria nella popolazione mondiale, la continua circolazione del virus stesso sarà associata alla comparsa di nuove mutazioni e varianti. Con paesi del mondo in cui la copertura vaccinale è subottimale o virtualmente assente, come in alcune nazioni del Sud del mondo, il rischio di insorgenza di una variante completamente immunoeludente è, ad oggi, concreto. Con i viaggi e i contatti in tempo reale fra regioni lontanissime, qualsiasi punto del mondo può essere raggiunto in tempi rapidissimi da una variante emersa nel più remoto sito del pianeta. Questo rischio può essere mitigato da una parte rallentando in maniera drastica la circolazione del virus, e dall’altra mantenendo elevata la reattività del SISTEMA nell’adattare la risposta all’epidemia (resilienza). Al riguardo, due capisaldi sono di primaria importanza: 1. il mantenimento di livelli costanti ed elevati di sequenziamento genomico a livello globale, per seguire ed eventualmente anticipare l’evoluzione del virus e fornire così gli elementi per decidere quando e come adeguare la composizione del vaccino, e 2. lo sforzo di copertura di tutta la popolazione mondiale con il vaccino che è disponibile al momento, eventualmente coadiuvato da somministrazioni di richiamo, che, pur essendo formulato contro la variante originale del virus, è tuttora in grado di garantire una significativa protezione contro le VOC al momento circolanti.


{kind=link}